-
法规背景
《中华人民共和国药品管理法》
《中华人民共和国药品管理法实施条例》
《药品注册管理办法》
《关于进口药品再注册有关事项的公告》
《进口药材管理办法(试行)》
主管机关
国家药品监督管理局(原国家食品药品监督管理局)
申请人资质
境内申请人应当是在中国境内合法登记并能独立承担民事责任的机构,境外申请人应当是境外合法制药厂商。境外申请人办理进口药品注册,应当由其驻中国境内的办事机构或者由其委托的中国境内代理机构办理。
办理药品注册申请事务的人员应当具有相应的专业知识,熟悉药品注册的法律、法规及技术要求。
法规要求
申请进口的药品,必须获得境外制药厂商所在生产国家或者地区的上市许可;未在生产国家或者地区获得上市许可,但经国家食品药品监督管理局确认该药品安全、有效而且临床需要的,可以批准进口。申请进口的药品应当符合所在国家或者地区药品生产质量管理规范及中国《药品生产质量管理规范》的要求。
申请材料分类及要求
(一) 进口中药、天然药物临床试验批准及注册证核发,注册分类及申报资料要求,参见《药品注册管理办法》。
(二) 进口化学药品临床试验批准及注册证核发,注册分类及申报资料要求,参见《药品注册管理办法》、《总局关于发布化学药品注册分类改革工作方案的公告》(2016 年第51 号)及《总局关于发布化学药品新注册分类申报资料要求(试行)的通告》(2016 年 第80 号)。
(三) 进口生物制品临床试验批准及注册证核发(含治疗用生物制品及预防用生物制品),注册分类及申报资料要求,参见《药品注册管理办法》。
(四) 临时进口药品审批申报资料要求,参见《关于进口药品再注册有关事项的公告》。
(五) 进口药材批件核发(含首次、非首次)申报资料要求,参见《进口药材管理办法(试行)》。
补充申请
新药申请、仿制药申请或者进口药品申请经批准后,改变、增加或者取消原批准事项或者内容的注册申请。
再注册申请
药品批准证明文件有效期满后申请人拟继续生产或者进口该药品的注册申请。
技术评审
技术审评工作时间按照下列规定执行:(一) 新药临床试验:90日;获准进入特殊审批程序的品种:80日;
(二) 新药生产:150日;获准进入特殊审批程序的品种:120日;
(三) 对已上市药品改变剂型和仿制药的申请:160日;
(四) 需要进行技术审评的补充申请:40日。
监测期
监测期自新药批准生产之日起计算,最长不得超过5年。
监测期内的新药,国家食品药品监督管理局不批准其他企业生产、改变剂型和进口。
有下列情形之一的药品不予再注册:
(一) 有效期届满前未提出再注册申请的;
(二) 未达到国家食品药品监督管理局批准上市时提出的有关要求的;
(三) 未按照要求完成IV期临床试验的;
(四) 未按照规定进行药品不良反应监测的;
(五) 经国家食品药品监督管理局再评价属于疗效不确、不良反应大或者其他原因危害人体健康的;
(六) 按照《药品管理法》的规定应当撤销药品批准证明文件的;
(七) 不具备《药品管理法》规定的生产条件的;
(八) 未按规定履行监测期责任的;
(九) 其他不符合有关规定的情形。
我们的服务
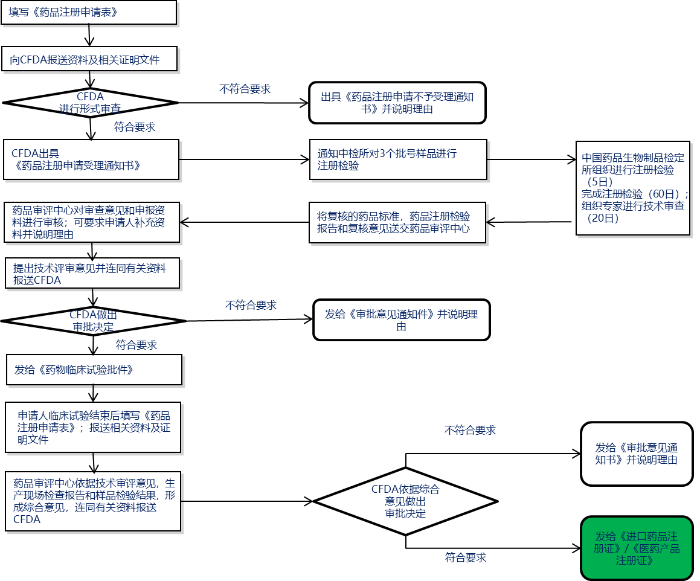
申报流程